1. The original intention of PhaSePred
Phase separation (PS) is an important mechanism that mediates the compartmentalization of proteins in cells.
Multivalent weak interactions between these molecules are the driving force of PS. The interactions can
generally be classified into two categories: one mediated by intrinsically disordered regions (IDRs) and the
other mediated by multiple modular domains or motifs. A difference between the two PS mechanisms is that a
single molecule species can undergo IDR-mediated phase separation, while phase separation mediated by
multiple interacting domains often involves two or more different molecule species. Herein, we characterize
proteins that can self-assemble to form condensates as self-assembling phase-separating (PS-Self) proteins,
and we define proteins whose phase separation behaviors are regulated by partner components (proteins or
nucleic acids) as partner-dependent phase-separating (PS-Part) proteins.
Currently, there is no computational tool that can identify partner-dependent phase-separating proteins.
However, most phase-separating systems involve multiple partners in biological conditions. A similar pattern
was displayed in the annotations collected from PhaSepDB with more PS-Part than PS-Self proteins. Therefore,
the screening of potential partner-dependent proteins is of great importance.
Sequence-based analysis tools are commonly used to screen phase-separating proteins. However, current PS
predictors recognize vastly different kinds of proteins because they were developed to screen various
sequence features, calling for the development of a comprehensive predictor.
Herein we build PhaSePred, a meta-predictor that incorporates prediction scores of multiple PS-related
predictors, including the self-assembling phase-separating predictor SaPS, the partner-dependent
phase-separating predictor PdPS, granule-forming propensity predictor catGRANULE, prion-like domain (PLD)
predictor PLAAC, π-contact predictor PScore, IDR predictor ESpritz, low-complexity region (LCR) predictor
SEG, hydropathy prediction from CIDER, coiled-coil domain predictor DeepCoil, immunofluorescence (IF)
image-based droplet-forming propensity predictor DeepPhase.
2. Construction of self-assembling and partner-dependent phase-separating predictors
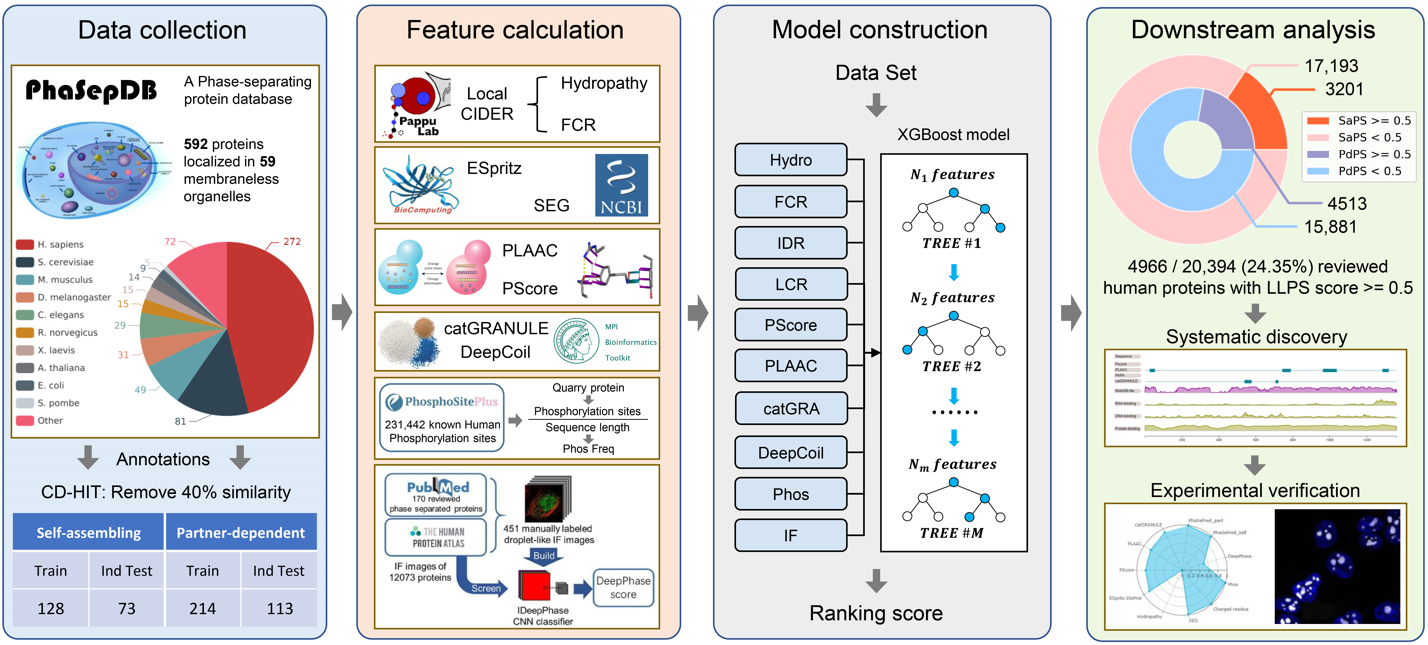
The process of constructing SaPS and PdPS models can be divided into three steps:
1) Data collection:
Proteins used to train the self-assembling and partner-dependent phase-separating predictors were collected
from PhaSepDB 2.0. The annotated proteins in PhaSepDB 2.0 were divided into two groups: PS-self and
PS-other. 'PS-self' refers to those proteins that can undergo self-assembling phase separation in vitro.
'PS-other' refers to those proteins contributing to the formation of biomolecular condensates. If a protein
participates in an MLO with partner components, its partners will be recorded in the 'Partner' column. We
selected proteins labeled 'PS-self' as self-assembling phase-separating proteins, and selected proteins with
the protein or nucleic acid partner as our defined partner-dependent proteins.
2) Feature calculation:
Features with batch prediction or provide predicted results were used to train the model. The Hydropathy and
FCR score of a protein was calculated by localCIDER using the default parameter. The Hydropathy score was
defined as the average hydropathy of each residue from a normalized Kyte–Doolittle hydrophobicity scale, and
the FCR score was calculated by dividing the total number of D, E, R, K residues by the sequence length. We
used the ESpritz DisProt program with the decision threshold set at a 5% false positive rate (FPR) to
predict potential disordered regions, and we used the SEG local package with default parameters to detect
low-complexity regions within a given protein sequence. The fraction of IDR or LCR was defined as the number
of amino acids in the corresponding domain divided by the sequence length. Each protein's PScore, PLAAC, and catGRANULE score was calculated using the corresponding tools under the default parameters. However, PLAAC
provides three summary scores for a given sequence, including LLR, CORE, and PRD. Since the LLR score is
more appropriate in whole-proteome screening, we used the normalized LLR score (NLLR) to represent the
PLD-forming propensity. The Python package DeepCoil was used to detect potential coiled-coil (CC) structures. We
used 0.82 as the threshold for CC structure detection and changed the score to 0 and 1 to indicate whether a
protein contains the predicted CC structure. The phosphorylation sites of human proteins were downloaded
from PhosphoSitePlus (retrieved Sep 8, 2020). The Phos frequency was defined as the number of
phosphorylation sites divided by the protein sequence length. IF image-based droplet forming propensities
were collected from Supplementary Table 2 of DeepPhase. We submitted 12,073 human Ensembl gene IDs provided
by DeepPhase to UniProtKB and retrieved 11,982 UniProt entries for further analysis.
3) Model construction:
Incorporating the features mentioned above, we used the Python package of XGBoost to construct models for
predicting self-assembling phase-separating (SaPS) and Partner-dependent phase-separating (PdPS) proteins,
respectively. Models with 8 features (Hydropathy, FCR, IDR, LCR, PScore, PLAAC, catGRANULE, DeepCoil) are
built on all-species data, while models with 10 features (the 8 features described above plus Phos frequency
and DeepPhase) are only built on the human proteome.
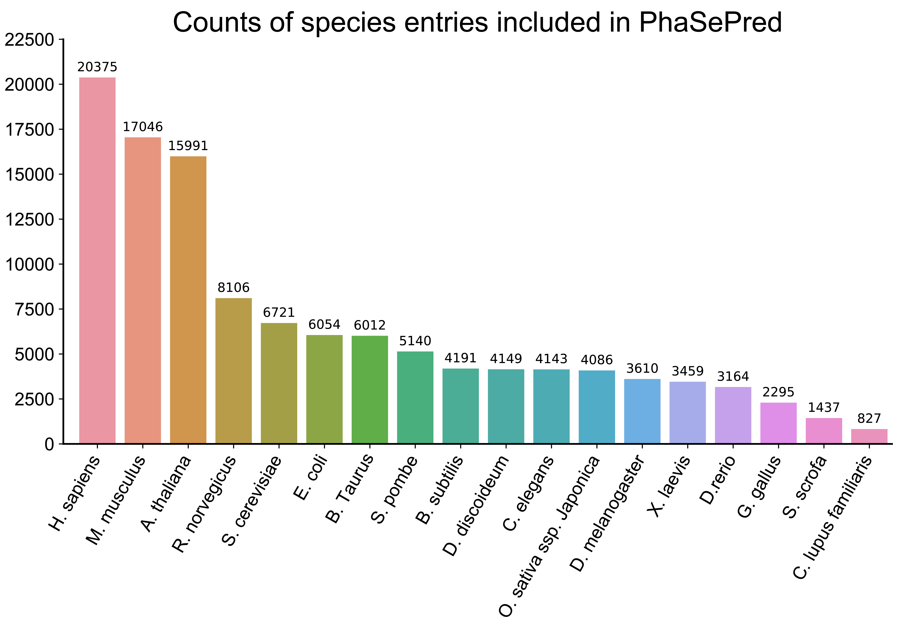
3. Data distribution
Currently, PhaSePred contains UniProt proteins from 18 well-studied species, including Homo sapiens (human),
Mus musculus (mouse), Arabidopsis thaliana (mouse-ear cress), Rattus norvegicus (rat), Saccharomyces
cerevisiae
(baker's yeast), Escherichia coli (strain K12 / DH10B), Bos taurus (bovine), Schizosaccharomyces pombe
(strain 972 / ATCC 24843) (fission yeast), Bacillus subtilis (strain 168), Dictyostelium discoideum (slime
mold),
Caenorhabditis elegans, Oryza sativa subsp. japonica (rice), Drosophila melanogaster (fruit fly), Xenopus
laevis (African clawed frog), Danio rerio (zebrafish) (Brachydanio rerio), Gallus gallus (chicken), Sus
scrofa
(pig), and Canis lupus familiaris (dog) (Canis familiaris).
As of August 2021, integrated predictions for 116,806 sequences were available.
4. Prediction of LLPS-related features
We provided residue-level scores and/or annotations of eleven LLPS-related features:
1) The domain information was annotated by the local package of InterProScan with the following command:
./interproscan.sh -i /inputFilePath/source.fasta -o /outputFilePath/target.tsv -f tsv
We only kept the annotation information from the Pfam database.
2) The granule-forming propensity was predicted by the web server of catGRANULE with default parameters.
3) The prion-like domains (PLDs) were predicted by the local package of PLAAC with the following
command:
java -jar ./plaac.jar -i /inputFilePath/source.fasta -a 1 -p all
PLAAC provides three summary scores for a given sequence, including LLR, CORE, and PRD. Since the LLR score
is more appreciated in whole-proteome screening, we used the normalized LLR score (NLLR) to represent the
PLD-forming propensity.
4) The π-contact propensities were predicted by the local package of PScore with the following command:
./PScore.py /inputFilePath/source.fasta -output /outputFilePath/target.out -mute
-overwrite -residue_scores -score_components
5) The intrinsically disordered regions (IDRs) were predicted by the local package of ESpritz with the
following command:
./espritz.pl /inputFilePath D 0
We set the prediction type at 'Disprot' and the decision threshold at '5% false-positive rate (FPR)'.
6) The residue hydropathy was predicted by the Python package of localCIDER. We used the function
'get_linear_hydropathy()' with the default parameter, which returns the local hydropathy with the
Kyte-Doolite hydropathy scale.
7) The coiled-coil (CC) structures were predicted by the Python package of DeepCoil with GPU acceleration.
We used the column 'cc' to represent the coiled-coil propensity, which ranges from 0 to 1.
8) The low-complexity regions (LCRs) were predicted by the local package of SEG with the following
command:
./seg /inputFilePath/source.fasta -x
9) The fraction of charged residues (FCR) was counted using the Python package of localCIDER. We used the
function 'get_FCR()' with the default parameter, which assumes a neutral pH where only R/K/D/E are
charged.
10) The Phosphorylation sites were collected from the PhosphoSitePlus (fetched Sep 8, 2020). This property
was only available on the Human proteome.
11) The immunofluorescence (IF) image-based droplet-forming propensity was collected from DeepPhase. This
property was only available on the Human proteome.
5. Related links
| PhaSepDB | The database of phase-separation related proteins |
|---|---|
| MloDisDB | A manually curated DataBase of the relations between MembraneLess Organelles and DISeases |
| PhaSePro | PhaSePro is the comprehensive database of proteins driving liquid-liquid phase separation (LLPS) in living cells |
| DrLLPS | Data resource of liquid-liquid phase separation |
| LLPSDB | A database of proteins undergoing liquid-liquid phase separation in vitro |
| PLAAC | PLAAC searches protein sequences to identify probable prion subsequences using a hidden-Markov model (HMM) algorithm. |
| PSPer | Unsupervised prediction of proteins able to form phase-separated liquid droplets acting as membraneless organelles. |
| Pscore | Pi-Pi contacts are an overlooked protein feature relevant to phase separation. |
| catGRANULE | catGRANULE is an algorithm to predict liquid-liquid phase separation propensity (LLPS). |
| R+Y | Critical concentration prediction based on number of arginine and tyrosine residues, extrapolated from FET family proteins. |
| MobiDB | a database of protein disorder and mobility annotations. |
| DeepCoil | a fast and accurate prediction of coiled-coil domains in protein sequences. |
| localCIDRE | resources to analyze sequence-ensemble relationships of intrinsically disordered proteins. |
| Espritz | Accurate and fast prediction of protein disorder. |
| SEG | Statistics of local complexity in amino acid sequences and sequence databases. |
| PhosphoSitePlus | provides comprehensive information and tools for the study of protein post-translational modifications (PTMs) including phosphorylation, acetylation, and more. |
| DeepPhase | Proteome-scale analysis of phase-separated proteins in immunofluorescence images. |
| The Human Protein Atlas | The Human Protein Atlas is a Swedish-based program initiated in 2003 with the aim to map all the human proteins in cells, tissues and organs using integration of various omics technologies. |